
Ⅰ. Title
염색체 DNA의 정제 및 분석
Ⅱ. Purpose
1. 대장균에서 염색체 DNA를 분리, 정제하는 과정에서 시약과 효소의 작용 이해
2. PCR과 전기영동의 사용방법 및 원리 익히기
Ⅲ. Theory
1. 유전물질의 구조
세포를 이용해서 실험을 하는 경우, 다루기 쉬우며 배양이 쉬운 세포를 사용한다. 효모, 예쁜꼬마선충(caenorhabitis elegans) 등을 많이 사용하지만, 대장균 (E. coli)이 많이 사용된다. 이와 같은 세균에는 염색체 DNA(Bacterial DNA)와 plasmid를 갖고 있다. 따라서 해당 세포의 유전자를 실험에서 사용한다면, Bacterial DNA와 plasmid를 구분해야 한다.
1) Bacterial DNA - 뉴클레오타이드와 DNA의 구조와 특성
세포 속에는 유전 물질이 저장되어 있다. 그 중에서 DNA는 핵산의 한 종류로 인산, 당 그리고 염기(base)로 구성된 뉴클레오타이드(nucleotide)의 복합체이다. DNA 구조를 이해하기 위해서 뉴클레오타이드의 구조를 살펴봐야 한다.
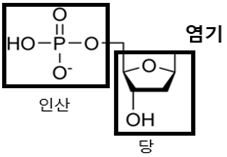
뉴클레오타이드의 구조는 다음과 같다. 뉴클레오타이드는 인산과 당 그리고 염기가 각각 1:1:1로 구성되어 있다. 우선, 당은 탄소 5개로 구성된 5탄당이다. 당의 탄소는 산소를 기준으로 시계방향으로 순서를 매겨, 3번 탄소에 수산화기가 연결되며, 5번 탄소에 인산이 연결된다. DNA를 구성하는 당은 디옥시리보오스다. 또한, 인산은 DNA를 구성할 때 공유결합을 통해 외부 골격을 형성한다. 이 과정에서 인산은 수소원자를 내어놓기 때문에 전기적으로 (-) 부호를 띤다. 마지막으로 염기의 종류로는 사이토신(C), 구아닌(G), 아데닌(A) 그리고 티민(T)이 있다.

NA는 염기 사이의 수소결합과 인산-디옥시리보오스 공유 결합을 통해서 DNA 구조를 형성한다. 염기는 상보적 수소 결합으로 서로 결합을 한다. 아데닌과 티민은 2개의 수소결합을 통해서 퓨린계를 형성한다. 구아닌과 사이토신은 3개의 수소결합을 하며, 피리미딘계를 형성한다. 그리고 인산과 당의 결합 구조는 탄소의 번호를 이용해서 설명한다. 인산에서 수소가 분리되어 3번 탄소와 연결된다. 인산과 당의 결합으로 형성된 외부 골격을 5번 탄소에서 시작하여, 3번 탄소에서 끝난다고 말한다.

편, DNA는 구조에 따라서 3가지 DNA로 구분할 수 있다. 왼쪽의 그림에서 왼쪽부터 차례대로 A-DNA, B-DNA, 그리고 Z-DNA이다. 우리에게 익숙한 DNA는 가운데의 B-DNA이며 10 bp를 갖는다. 다만, A-DNA는 나선의 기울기가 30°가량 기울어져 있으며, B형보다 더 회전 정도가 심해 한 나선에 11 bp를 갖는다. Z형은 B형과 거울상 대칭을 보이며 12bp를 갖는다.
2) 플라스미드(plasmid)의 구조와 특성
plasmid는 고리 형태를 보이는 유전물질이며, bacterial DNA와는 다르게 스스로 복제할 수 있다. 일반적으로 plasmid는 supercoiled form으로 존재한다. 이는 plasmid가 두 가닥으로 구성이 되어 있다. 정제 등 plasmid에 외부 요인이 관여한다면 3가지 형태를 가질 수 있다.
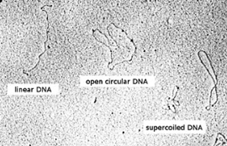
외적 요인의 존재에도 불구하고 변형이 가해지지 않을 때에는 supercoiled DNA, 두 가닥 중 한 가닥이 잘릴 때 관찰되는 open-circular DNA (Nicked DNA), 그리고 두 가닥이 모두 끊여졌을 때에는 liner DNA 형태가 관찰된다.
2. DNA의 분리
박테리아는 크기가 작으며, 그 구조가 단순하다. 특히 원핵세포의 경우 핵이 따로 존재하지 않기 때문에 세포의 내부와 외부를 구분하기 위해서 세포막과 세포벽이 있다. 다만, 이러한 형태로 존재하는 경우 세포 내부에 존재하는 물질을 이용하고 분석하기 쉽지 않아서 DNA 분리 과정을 거쳐야 한다. DNA 분리는 세포벽과 세포막을 분해하여 유전물질을 얻어내는 것이다. 세포벽은 그램양성균또는 그램음성균으로 구성되어 있으며, 세포막은 인지질과 단백질 등으로 구성이 되어있다. 다양한 방법이 존재할 수 있으나 Alkaline lysis 방법을 많이 사용한다. Alkaline lysis 방법은 높은 pH를 알칼리성 음이온 계면활성제 용액을 이용해서 대장균 세포를 용해시킨 후 원심분리 방법을 이용해서 plasmid DNA와 bacterial DNA (염색체 DNA)를 구분할 수 있게 된다.

(1) 단계는 주어진 박테리아의 세포벽을 분리하는 단계이다. 이를 위해서 이온성 계면활성제 EDTA와 효소 lysozyme을 사용한다. EDTA는 마그네슘 이온과 칼슘 이온과 같은 2가 양이온들과 chelating 하는 성격을 갖고 있다, 세포벽에는 칼슘 이온이 있으며, 이온은 세포벽이 잘 유지될 수 있도록 한다. EDTA를 이용하면 세포벽 사이의 칼슘 이온이 분리되어 박테리아가 잘 분해될 수 있도록 한다. 한편, 원핵세포의 세포벽은 펩티도글리칸(peptidoglycan)으로 구성되어 있으며, 이 당은 1,4-glucosidic linkage로 구성이 되어 있다. lysozyme은 1,4-glucosidic linkage를 분해하여 세포를 용해되도록 한다. (1) 과정에서 넣어주는 sucrose는 분해하고자 하는 세포와 EDTA + lysozyme 등으로 구성된 용액 사이의 삼투압을 유지하는 용도로 이용된다. 생체물질은 pH에 민감하여 변성이 쉽게 발생한다. (2) 단계에서는 NaOH 등 강염기를 혼합하여 Bacterial DNA와 plasmid의 변성을 일으킨다, 이 과정에서 Bacterial DNA는 구조의 크기가 커 변성이 많이 발생하며, plasmid는 이보다는 변성이 적게 일어난다. 또한, Triton X-100 계면활성제를 이용하여 인지질의 소수성 부분을 녹이고 단백질은 변형시켜 세포막을 용해한다. (3) 단계에서는 (2) 단계를 거쳐 pH가 매우 높은 염기성 상태의 용액을 다시 중성상태로 만든다. 이 과정에서 plasmid는 변성정도가 적어 원래의 형태로 돌아올 수 있으나 Bacterial DNA는 그렇지 못한다. 따라서 분획을 이용하면 plasmid와 세포 찌꺼기, 변성된 단백질 등과 얽힌 변성된 bacterial DNA가 분리된다.
3. PCR (Polymerase Chain Reaction)
일반적으로 생물 내에서의 메커니즘 만을 이용하여 분석하고자 하는 유전 물질을 얻어내려면 시간 이 오래 걸린다. (in vivo) 하지만 세포 밖에서 체내에서 유전 물질은 복제하는 메커니즘을 모방하는 방식을 이용한다면 비교적 빠른 시간 내에 분석하고자 하는 물질을 얻어낼 수 있다. (in vitro) 이 방법을 PCR (Polymerase Chain Reaction, 중합효소연쇄반응) 이라고 한다.
1) 구성 요소
PCR을 진행하기 위해서 복제하고자 하는 물질과 함께 아래의 4가지 물질을 같이 넣고 복제를 진행해야 한다.
① DNA Template
DNA Template는 실험에서 사용하고자 하는 부분을 말한다. 즉, PCR cycle을 통해서 증폭하고자 하는 표적 염기서열을 말한다. 앞서 살펴보았듯이 Bacterial-DNA는 이중 나선 구조를 보이며 염기 A와 T, G와 C가 각각 수소 결합을 통해서 상보적인 염기서열을 갖고 있다. 한편, 방향성에 따라 anti-parallel한 결합을 보이기도 한다.
② Primer

생체 DNA를 복제하는 과정 중 primer가 붙어야만 복제가 시작된다. 즉, primer의 부착은 복제의 시작을 말하며 복제하고자 하는 부위를 결정한다. 또한 DNA 구조에서 인산기의 수산화기와 디옥시리보스의 3번 탄소와 연결이 되어 있기 때문에 복제 과정에서도 해당 결합을 필요로 한다. DNA polymerase도 마찬가지로 중합을 진행할 때 3번 탄소에 -OH를 제공해야 하기 때문에 primer는 필수적이다. primer의 길이는 다양하나 복제하고자 하는 DNA에 따라 적당한 길이를 설정해야 한다.
③ DNA Polymerase
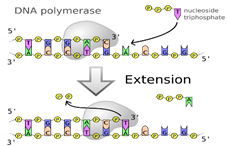
중합의 기본이 되는 template와 primer가 결합하면 DNA polymerase가 중합반응을 진행할 수 있다. 이 효소는 뉴클레오타이드를 연결하여 DNA를 복제한다. 복제의 방향은 DNA template에서 3번 쪽에서 5번 쪽으로 진행한다. 이는 상보적 결합에 의하여 primer의 5번 탄소에서부터 결합을 시작하기 때문이다. DNA polymerase의 종류는 많으나, PCR은 비교적 고온에서 진행되는 경우가 많다. 그래서 뜨거운 온천에서 생존하는 세균 Thermus aquaticus으로부터 얻은 Taq polymerase를 많이 이용한다.
④ Nucleotide
DNA polymerase가 DNA를 연장하려면 결합에 필요한 뉴클레오타이드가 필요하다. template의 염기 서열과 상보적으로 결합이 진행된다.
2) PCR cycle
PCR 메커니즘은 온도 정보의 변화를 따라 3단계로 구분된다.

① DNA 변성 (Denaturation)
이 과정은 target DNA (gene of interest)를 포함한 DNA를 고온으로 가열하는 과정이다. 단백질 등에 고온이 가해지면 변성이 올 수 있는데, DNA도 마찬가지로 변성이 진행된다. 고온 상황에서는 DNA의 수소결합이 더 이상 형태를 유지하지 못하고 끊어진다. 이 과정을 통해 이중 나선 구조(dsDNA)를 보이던 DNA가 단일 가닥 형태(ssDNA)로 벌려진다. 높은 온도일수록 denaturation이 잘 일어나지만, DNA polymerase 등 다른 물질도 변성이 진행될 수 있어 적당한 90℃ 정도에서 진행한다.
② 프라이머의 결합 (Annealing)
DNA polymerase가 중합반응을 진행하려면 primer가 변성이 일어난 template DNA에 붙어야 하기 때문에 온도를 어느정도 낮추어야 한다. 온도를 많이 낮추면 염기들이 상보적으로 결합하지 못하고 이상한 결합이 발생할 수 있기 때문에 결합에 적당한 온도를 찾아야 한다. 이 온도는 프라이머에 포함되어 있는 염기의 개수에 의해 결정된다. 프라이머의 길이에 따라서 Annealing 과정에서 설정되어야 하는 적당한 온도는 아래와 같이 구할 수 있다.

다만, 이 식은 다양한 환경 요인(중합효소의 종류, 농도 등)의 영향력 아래에 있기 때문에 각 상황에 맞는 을 계산해주는 프로그램을 이용하는 경우가 많다. 한편, 구한 보다 약간 더 낮은 온도에서 Annealing을 진행하면 더 결합이 잘된다.
③ DNA 합성 (Elongation)
DNA polymerase를 이용해서 DNA를 복제하는 과정이다. 프라이머를 기준으로 5번 탄소에서 3번 탄소 방향으로 합성이 진행된다, 이때에는 template를 기준으로 상보적인 염기 서열을 가진 DNA가 복제가 된다.
해당 cycle을 반복적으로 진행하면 우리가 얻고자 하는 DNA 물질을 많이 복제할 수 있다. 다만 생성된 물질은 바로 사용하지 못하며, 정제 과정을 거쳐야 사용할 수 있다.
4. 전기영동
전기영동을 이용하면 생체 고분자의 특성을 분석하고 정제할 수 있다. 전기영동은 전기장에 걸려있는 분석 및 정제하고자 하는 물질이 전하를 띨 수 있는 성질을 이용한다. 특히 DNA는 인산-당 결합으로 구성된 외부 골격이 (-)를 전하를 띠기 때문에 분석에 용이하다.
1) 이동 속도 결정
전기 영동에서 DNA들은 몇 가지 특성에 의하여 이동하는 속도와 거리가 결정된다. DNA의 net charge, DNA의 크기 및 모양, 전기장의 세기, supporting meduium의 성질, 그리고 전기영동이 진행되는 온도에 의하여 결정된다. 분석되는 물질을 구형이라고 가정하고 나비에-스토크스 방정식을 이용하면 아래와 같은 식을 얻을 수 있다.

(두 전극의 전위차: E, 전극 사이의 거리 d, 분자의 전하 q, 용액의 점성도: , 반지름: , 물질이 움직이는 속도: )
한 번의 전기 영동을 실시하면 sample들은 같은 시간동안 분석이 되므로 이동 속도가 더 빠르면 더 많은 거리를 이동한다고 생각할 수 있다.
전극 사이의 전압과 물질의 전하와 더 많은 거리를 이동할 수 있다. 반면, 물질의 크기가 클수록 (즉, 물질의 부피가 클수록), buffer의 점성도가 클수록, 두 전극 사이의 거리가 클수록 물질은 더 적은 거리를 이동한다. 또한, 전기영동이 시행되는 온도가 높을수록 더 빠르게 이동할 수 있으나, 너무 높은 온도는 분석 물질의 변형을 유발할 수 있으므로 주의해야 한다.
한편, 전기영동을 진행할 때 물체의 속도가 무한하게 증가하지는 않는다. 전하를 띤 입자는 supporting medium을 통해 이동할 때 마찰력에 의해서 진행방향과 반대 방향으로 영향을 받기 때문이다.
2) 아가로오스 겔 전기영동 (Agarose gel Electrophoresis)
전기영동에는 다양한 종류가 있으나 agarose gel 전기영동에 대해서 자세히 살펴보도록 하자.

agarose는 D-galctos와 3,6-anhydro L-galactose가 결합되어 형성된 중합체다. 이 물질의 구조에서 보이는 구멍의 크기는 일정하지 않다. 그래서 분리 시 해상도가 좋은 편은 아니나, 넓은 범위의 bp 길이를 갖는 DNA 들을 분리해낼 수 있으며 다루기 쉽기 때문에 맣이 사용되는 전기영동법이다. agarose가 gel 형태로 만들어져 있지 않다면 전기영동시 사용할 buffer에 녹여서 gel 형태로 굳혀 사용하며 그렇지 않은 경우에는 제조된 gel의 구성에 따라 buffer를 사용하는 것이 좋다.
ⅰ) agarose gel 전기영동에서의 이동 속도에 영향을 주는 요인
(ㄱ) gel 사이의 구멍을 통과해야 DNA가 이동할 수 있다. 그렇기에 크기가 비교적 작은 크기의 DNA가 더 많이 움직일 수 있다.
(ㄴ) agarose gel의 농도에 따라서 분리할 수 있는 DNA의 크기가 달라진다.

(ㄷ) 비슷한 분자량의 DNA를 분리하더라도 분리하는 DNA의 형태에 따라서 이동하는 속도가 달라질 수 있다. 앞서 살펴보았던 plasmid는 supercoiled circular, nicked circular, linear형태로 존재할 수 있다. buffer로 EtBr을 사용하는 경우 일반적으로 supercoiled circular plasmid가 다른 plasmid보다 빨리 움직일 수 있다.
(ㄹ) buffer의 이온강도에 의해서도 물질의 이동이 영향을 받는다. 이온 강도는 용액 안에 들어있는 이온 농도를 표현하는 수치인데, 이 값이 작으면 DNA 이동 속도가 매우 느리다. 그러나 지나치게 이온 강도가 센 용액을 사용하면 전류 때문에 고온이 발생할 수 있으며 이는 시료의 변성을 유도하고 gel을 녹일 수 있기 때문에 주의해야 한다.
ⅱ) buffer
agarose gel 전기영동을 진행할 때 2가지 종류의 buffer를 이용한다. 각 buffer의 조성과 특징을 아래의 표로 정리했다.
|
|
TAE (Tris-acetate-EDTA) buffer |
TBE (Tris-Boric acid-EDTA) buffer |
|
구성 |
Tris base, Acetic acid, EDTA |
Tris base, Boric acid, EDTA |
|
기능 |
Tris base: DNA를 이동시킬 수 있는 양이온 제공 EDTA: 금속이온(DNase의 양이온 Cofactor)제거 Acetic acid: pH 조절 Boric acid: 효소의 작용 억제 가능 |
|
|
사용 대상 |
비교적 긴 DNA 분석에 적합 (>12kb) |
비교적 짧은 DNA 분석에 적합 (<2kb) |
한편, polyacrylamide gel을 이용한 전기영동도 널리 이용된다. agarose gel보다 gel의 합성이 어려우나, 더 많은 양의 시료를 더 뚜렷하게 정제할 수 있다는 장점이 존재한다.
ⅲ) Gel staining dye

일반적이 DNA의 색은 무색으로, 이를 관찰하라면 또 다른 처리가 필요하다. 이를 위해 사용하는 것을 Gel Staining Dye라고 한다. 이 물질들은 염기 사이의 수소 결합에 작용한다. 처리 후 자외선을 이용하면 형광 정도가 기존보다 더 증가해서 관찰이 용이하다. 대표적인 예시로는 EtBr이 있다. 다만, 이 물질은 독성이며 DNA에 직접적인 영향을 줄 수 있어 RedSafe 등 다른 시료로 대체하여 사용 가능하다.
Ⅳ. Procedure
1. PCR
- E coli(대장균)의 gDNA에서 gldA(1104bp), mgsA(459bp) 유전자를 증폭
1) PCR premix tube에 gldA, mgsA를 추출할 각각의 시약 넣기
- Rnase free water (14μL) + E coli (2μL) + gldA upstream primer (2μL)
+ gldA downstream primer (2μL)
- Rnase free water (14μL) + E coli (2μL) + mgsA upstream primer (2μL)
+ mgsA downstream primer (2μL)
-> 2개의 tube 만들기
2) PCR 기계에 tube를 꽂은 뒤 온도와 cycle을 설정하여 작동
|
과정 |
온도 (℃) |
시간 |
주기 (cycle) |
|
|
Initial Denaturation |
98 |
10분 |
1 |
|
|
Denaturation |
98 |
10초 |
30 |
|
|
Annealing |
mgsA |
59 |
30초 |
|
|
gldA |
55 |
|||
|
Extension |
72 |
1분 |
||
|
Final Extension |
72 |
10분 |
1 |
|
표 SEQ 표 \* ARABIC 3 PCR cycle에서 필요한 온도와 실행 주기 횟수
한편, 온도를 gradient로 설정하여 왼쪽 5칸에 mgsA, 오른쪽 4칸에 gldA를 두기
2. 전기영동
- 증폭된 DNA를 분리하여 확인
1) Agarose gel을 전기영동기에 놓기
2) TAE buffer를 전기영동기에 gel이 잠길 정도로 붓기
3) 홈에 DNA ladder 10μL씩 홈에 각각 주입
4) 2개의 PCR product를 10μL
5) 100V로 30분 동안 전압 걸기
6) 전기영동기를 끄고 Agarose gel을 UV-VIS에 올려 band를 확인
Ⅴ. Data & Result

1. DNA ladder와의 비교를 통한 gldA와 mgsA의 bp 측정값 및 오차율
- 단순히 육안을 통한 비교가 아닌 geogbra 프로그램에서 y 좌표를 표현한 후, 가장 인접한 두 DNA ladder와의 내분 정도를 비교하여 최대한 구체적인 수치를 얻으려고 했다.
|
sample + ladder |
y 좌표 |
상대적 차이 |
sample + ladder |
y 좌표 |
상대적 차이 |
|
1200bp |
1.643 |
1.407 0.233 |
500bp |
0.254 |
0.188 0.152 |
|
gldA |
1.236 |
mgsA |
0.136 |
||
|
1000bp |
1.033 |
400bp |
-0.016 |

- 위에서 구한 수치들과 이론 전개 정도와 비교한 정도를 아래의 표 2에 정리하였다.
|
|
theorical bp |
experimental bp |
오차율 (%) |
비고 |
|
gldA |
1104 |
1028 |
6.88 |
덜 전개됨 |
|
mgsA |
459 |
445 |
3.05 |
덜 전개됨 |
Ⅵ. Discussion
PCR 과정은 분석하고자 하는 DNA sample의 중 특정 DNA의 양을 최대한 극대화하여 후에 분석 시 정확도와 관찰 정도를 증대하는데 도움이 되는 과정이다. PCR 과정은 denaturation - annealing - polymerization 과정을 따르고 일반적으로는 DNA template, primer, DNA polymerase, nucleotide를 넣어주었다. 다만 이번실험에서는 조금 다른 점이 있다. E coli를 바로 넣어주어 polymerase 등을 따로 넣는 점을 방지했다. 또한 이들을 섞기 위해서 Rnase free water를 넣어준다. 이는 가수분해효소가 없는 증류수를 의미하며, DNA와 효소 등이 분해되는 것을 방지하기 위함이다. 이번 실험에서 관찰되는 PCR cycle에 대해 살펴보자. 이번 실험에서는 gldA와 mgsA를 사용하는데, 이는 DNA이기 때문에 이중결합을 하고 있다. DNA 복제 과정에서 DNA는 효소에 의해서 단일 결합으로 잠시 분리된다. denaturation에서는 DNA가 고온에서 변성을 일으킨다는 특성을 이용해서 강제로 단일 결합 상태로 만든다. 이는 이중 결합을 유지하고 있던 수소 결합이 고온에서 끊어지기 때문에 가능하다. 다만 너무 높은 온도에서 진행하는 것은 E coli와 내부에 포함된 효소도 변성이 진행될 수 있기 때문에 98℃에서 진행했다. 두 번째로 annealing 단계를 진행했다. 이 단계는 넣어준 primer가 분리된 단일 가닥 DNA(ssDNA)에 부착되어 DNA 형성을 진행하는 단계이다. primer가 E coli 속의 DNA에 붙는 과정인데, 이 과정이 진행되려면 PCR 기기의 온도를 낮추어야 한다. primer와 ssDNA가 결합하는 것은 수소결합이 형성되는 것을 의미하기 때문이다. 온도가 낮을수록 수소 결합은 더 많이 형성될 것이나, 지나친 온도 저하는 undesired sample을 얻을 수 있기 때문에 각 primer마다 요구되는 annealing temperature Tm를 계산해야 한다. 계산 방법은 아래와 같다.

gldA와 mgsA의 upstream primer와 downstream primer의 염기서열과 요구되는 Tm을 아래의 표 3에 정리했다. 각 primer와 primer의 농도 가중치 정도를 반영하여 Tm을 결정할 수 있으며, 이 실험에서는 농도가 같은 시료를 사용하므로 Tmu와 Tmd의 평균값을 채택하면 된다.
|
|
gldA |
mgsA |
|
Upstream primer |
5’-ATGGAACTGACGACTCGCAC-3’ |
5’-ATGGACCGCATTATTCAATC-3’ |
|
Tmu (℃) |
4×(5+6)+2×(6+3)=62 |
4×(3+5)+2×(6+9)=56 |
|
Downstream primer |
5’-TTACTTCAGACGGTCCGCGA-3’ |
5’-TTATTCCCACTCTTGCAGGA-3’ |
|
Tmd (℃) |
4×(5+6)+2×(4+5)=62 |
4×(3+6)+2×(4+7)=58 |
|
Tm (℃) |
(62+62)/2=62 |
(56+58)/2=57.5 |
위의 결과는 이상적인 Tm이며 실제로는 더 나은 결합을 위해서 해당 값에서 약간 차이가 나는 온도에서 annealing을 진행하며, gldA의 경우 55℃ mgsA의 경우 59℃으로 Tm으로 설정했다. annealing 과정을 여러 번 진행해서 복제된 DNA sample을 얻는데 cycle의 수와 product는 마냥 선형적인 관계는 아니기 때문에 너무 많이 진행하여 부산물이 생성되지 않도록 주의해야 한다. 마지막으로 결합된 primer를 기준으로 DNA 연장을 진행하여 우리가 얻고자 하는 gldA와 mgsA를 얻는다 이는 E coli 내부에 있는 DNA polymerase가 관여한다. 더 나은 PCR 기기는 T gradient를 각 cell마다 설정할 수 있으나 실험에서 사용한 PCR 기기는 그렇지 못해서 각 DNA를 얻기 위해서 2번 같은 과정을 반복해야 한다. PCR 과정에서 template나 primer의 양이 너무 적으면 DNA와 primer의 결합이 잘 진행되지 않아서 PCR 과정을 진행한다고 하더라도 얻고자 하는 DNA가 잘 얻어지지 않을 수 있다. 그리고 소량의 물질을 다루기 때문에 피펫을 잘못 사용하여 벽면에 각 물질이 붙어버리거나 하면 마찬가지로 얻고자 하는 sample을 잘 얻을 수 없기에 이 부분을 유념해야 한다. 마찬가지로 소량의 물질을 다루기에 각 물질들이 오염되지 않은 상태에서 진행하도록 해야 한다. 예를 들어 대기 중 수분 속에 포함되어 있을 수 있는 가수분해 효소는 시료를 분해할 수 있다. 한편, 온도는 관성이 큰 특성이기에 온도 변화를 즉각적으로 조정하기 힘들어서 각 과정에서 요구하는 온도까지 도달하는데 시간이 걸리고, 이는 부정확한 product를 얻는데 관여할 수 있다. PCR 과정을 통해 얻은 product가 맞는지 확인하기 위해서 단백질 분석을 진행하기도 하며, 얻고자 하는 물질의 종류들을 아는 경우 전기영동을 이용해서 종류 결정 과정을 간소화할 수 있다.
일반적인 전기영동 실험에서는 우리가 실험을 진행하는 환경에 맞추어 제작할 필요가 있으나, 이번 실험에서는 특별히 그 과정을 진행하지 않고 Safeshine Agarose Gel을 사용했다. agarose gel과 전기영동 시 이용하는 TAE buffer가 들어가는 것은 일반적인 agarose gel의 제법과 다른 점이 없다. 다만 gel 자체에 gel staining dye 역할을 수행하는 Safeshine Green이 포함되어 있어서 PCR product를 running sample로 이용할 때 gel staining dye를 추가해야 하는 번거로움을 덜 수 있었다. 이 과정에서 product의 양이 매우 작기 때문에 dye와 혼합을 진행할 때 시료가 벽면에 붙는 등 손실을 최소화하기도 한다. 이번 실험에서 분리하고자 하는 DNA gldA와 mgsA의 bp 길이가 각각 1104 bp와 459 bp이기 때문에 해당 크기의 DNA를 잘 분리할 수 있는 1% agarose gel을 사용했다. 아래의 표 4는 DNA size를 잘 분리할 수 있는 agarose gel을 표로 나타낸 것이다.
|
Agarose gel (w/v %) |
DNA size 분리범위 (kb=1000bp) |
|
0.5 |
1 kb ~ 30 kb |
|
0.7 |
800 bp ~ 12 kb |
|
1.0 |
500 bp ~ 10 kb |
|
1.2 |
400 bp ~ 7 kb |
|
1.5 |
200 bp ~ 3 kb |
|
2.0 |
50 bp ~ 2 kb |
보다 DNA가 잘 이동할 수 있도록 TAE buffer를 사용했다. buffer는 pH의 범위를 일정하게 유지하도록 하는데, DNA가 pH 변화에 민감하다는 것을 고려하여 선정한 이동상이다. 실제로 running을 하면 DNA가 갖고 있는 (-) 전하 때문에 (+)극 쪽으로 이동을 할 수 있고, 이 이동의 driving force는 전기장에 걸린 전압이다. 정제를 마쳤다고 판단되면 UV-Vis 장치를 이용하여 전개정도를 비교한다. running 중 DNA의 수소결합에 관여한 safeshine green이 활성화되어 자외선을 발산하여 실제 전개된 정도를 비교할 수 있다.
다음으로 실험 결과를 살펴보자. 우선, UV-Vis 결과 DNA 끌림 현상이 관찰된다. DNA band 위 끌림 현상은 DNA의 농도가 높아 동시에 정제가 진행되지 못한 것을 의미하며, DNA band 아래 끌림 현상은 짧은 bp를 갖는 DNA 조각이 존재했기 때문에 관찰되는 현상이다. DNA 끌림이 발생했다고 판단되는 band는 mgsA이며 DNA band 아래 끌림이 관찰된 것으로 보아, PCR 과정 중 미처 결합을 하지 못한 DNA 조각이 있거나 전기영동 중 전기장에 영향으로 인해 일부 분해된 mgsA의 영향 때문일 수 있다. mgsA에 대해 DNA band 위 끌림도 관찰된 것으로 보이는 데, 실험 환경을 정확하게 알 수 없으나 사용한 agarose gel의 농도가 수용할 수 있는 분해 정도보다 더 높은 농도의 sample을 setting했거나 running 도중 safeshine green이 지나치게 관여하여 DNA 구조에 영향을 미쳤기 때문일 수도 있다. 한편, gldA에 대해서는 DNA 끌림은 관찰되지 않으나 100bp 부근에 미세한 band가 관찰된다. 이는 PCR 과정 중 미쳐 결합을 하지 못한 template가 running에 참여한 것으로 생각해볼 수 있다. 표 2를 살펴보자. 해당 표의 비고를 보면 두 DNA 모두 예상보다 전개가 덜 되었다고 판단된다. 이는 그림 1에서 DNA ladder를 봐도 알 수 있는데, 실제 전개된 DNA ladder에 비해서 DNA band들이 뚜렷한 간격을 유지하니 못하는 것으로부터 생각해볼 수 있다. 이 오차에 대한 대표적인 원인으로는 충분한 running 시간을 확보하지 못한 것이 있다.
PCR 증폭은 다양한 방면에서 사용되고 있는데, 대표적으로는 의학적 진단에 주로 사용된다. 종양 세포에 대해서 유전적으로 변화된 정도를 관찰하기도 하며 박테리아 및 바이러스 감염에도 사용된다. 최근 유행하고 있는 코로나 바이러스 진단 과정에서도 PCR 유전자 증폭 방법이 사용된다.
Ⅶ. Reference
1. 서강대학교 화공생명공학기초실험1 메뉴열 2020
2. 강호일, 전기영동 최신 프로토콜, 월드사이언스, 2006, pp. 14~18
'자료.zip > 화공생명공학전공실험' 카테고리의 다른 글
| 입도분석 (0) | 2021.02.01 |
|---|---|
| Hplc (1) | 2021.02.01 |
| FT-IR 분석 실험 (0) | 2021.02.01 |
| 접촉각 측정 (Contact Angle) (1) | 2020.12.17 |
| Grignard Reagent를 이용한 Triphenylmethanol synthesis 실험 (0) | 2020.10.28 |